Ab initio parameterization of van der Waals interactions
Ab initio parameterization of van der Waals interactions
Promotor(en): T. Verstraelen, D. Van Neck /MM_14_MODEV_02 / Model and software development, Many-particle physics, Nanoporous materialsThe van der Waals equation of state describes how the finite size and mutual attraction of particles determine the properties of non-ideal gases. At the mircoscopic scale, the van der Waals interaction is governed by an interplay between short-range Pauli repulsion and long-range attractive London dispersion. The relevance of these interactions goes far beyond the description of non-ideal gases. In any molecular simulation that involves physi- or chemisorption, the correct description of van der Waals interactions is crucial, e.g. for the adsorption, diffusion and catalysis in Metal-organic frameworks and zeolites, for the binding of pharmaceuticals to the active site of an enzyme, for the binding of organic substances on graphite and metal surfaces, and so on. It is also the force that geckos use to climb on smooth surfaces.
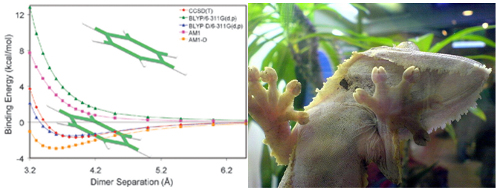
Figure: (left) Van der Waals interaction energy between two benzene molecules as function of the separation. (right) A gecko climbing up a glass window. The main challenge in the theoretical description of Van der Waals interactions is the dispersion term. It is the consequence of the correlated dynamics of distant electrons, which is notoriously difficult to compute accurately with quantum-mechanical techniques. To surmount this computational barrier, several dispersion-correction schemes for Density Functional Theory (DFT) were proposed over the past 10 years. The simplest approach is to add, for each pair of atoms, a pairwise interaction term of the form CR-6, where the parameter C is the strength of the dispersion interaction that is specific for the two atoms and R is the distance between the nuclei.
Simulations on extended systems (ordered porous materials, enzymes, …) are usually based on force-field simulations. In this technique, the computation of the electronic structure is replaced by empirical classical energy terms for the sake of computational efficiency. The van der Waals interaction is an important contribution in these models. Today, the DFT dispersion-correction schemes are not yet tested in the context of force fields.
The goal of this work is to develop a practical procedure to derive reliable van der Waals parameters based on DFT dispersion correction schemes. An initial procedure is already developed at the Center for Molecular Modeling and can be used as a starting point for this thesis. Several dispersion schemes are available in the literature that can be used for comparison and, if needed, these schemes may be further improved. The degree of empiricism varies: some schemes are based on experimental reference data while more advanced schemes solely rely on ab initio input. In order to obtain a broadly applicable procedure for the parameters, it is desirable to avoid any experimental input. The van der Waals parameters can be validated by a comparison with reference data:
- High-level ab initio computations (coupled-cluster, symmetry adapted perturbation theory, …) for small systems.
- High-level ab initio reference data taken from the literature for standard test sets, e.g. the S22 set.
- Experimental van der Waals parameters for small molecules.
This thesis has both computational (model validation) and theoretical (dispersion model development) aspects.

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Physics and Astronomy [CMFYST]KeywordsHost-guest interactions, Model development, DispersionRecommended coursesComputational physics (C001827); Many-body physics (C001759)
Contact
Dimitri Van Neck
Louis Vanduyfhuys
Toon Verstraelen