Stacking silver differently for improved surface properties
Stacking silver differently for improved surface properties
Promotor(en): V. Van Speybroeck, K. Lejaeghere /18SPEC07 / Nanoporous materials, SpectroscopyYou might think that little remains to be discovered about silver. It’s a noble metal with a close-packed fcc crystal structure, exhibiting a stacking sequence ABC ABC … along the (111) direction (also called 3C because it is cubic with a 3-element stacking motif). Due to its full d shell, relative rarity and excellent conductivity, it is used for a wide range of purposes, varying from jewellery to electronics. Like many other noble metals, silver is moreover an important catalyst: when adsorbed to its surface, some molecules react with each other more easily than they would without the silver support.
Recent investigations, however, showed that silver is not as simple as people thus far thought. Whereas the classical view is that silver assumes an fcc crystals structure, several polytypes have now been discovered that display substantially different properties. Polytypes are variations of a material that possess the same in-layer geometry, but which show a different stacking order. For silver, especially the 4H polytype has now attracted interest, which has a hexagonal crystal structure and displays an ABCB ABCB … stacking (see right figure). This polytype can be synthesized by electrodeposition or sputtering and differs significantly from 3C silver. This can be seen from its poorer metallicity, darker colour and higher hardness, for example.
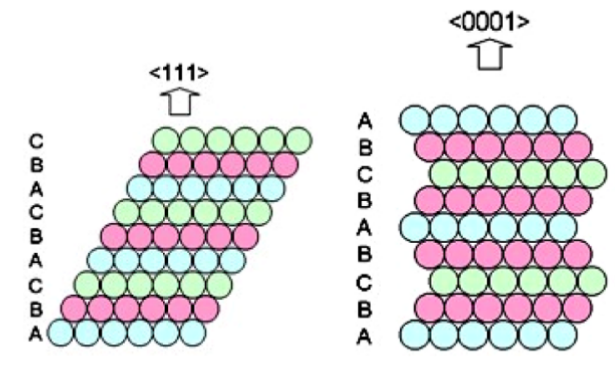
(from https://dx.doi.org/10.1088/0953-8984/26/2/025402)Not only the conductivity and mechanical properties of silver are changed by considering another polytype. Of particular interest to catalysis are the changes to the electronic structure. The differences between 3C and 4H silver suggest that the stacking order, and hence the polytype, can be modified to tune the position of the electronic bands and hence the interaction with redoxactive molecular species. However, little to nothing is known about the surface properties of silver polytypes. And that’s where we need your help.
Goal
The goal of this thesis is to model computationally how the stacking order of silver influences its surface properties. An obvious choice is to consider the [111] surface, perpendicular to the stacking direction, which will most likely be the predominant surface orientation. However, other crystal planes and surface defects (such as terraces, ledges and kinks) should be taken into account as well, since silver catalysts not only come in single-orientation surfaces but also as defected surfaces and nanoparticles, where a wider distribution of surfaces is present.
This thesis consists of two stages. On the one hand, we need to know the most basic traits of the different silver surfaces. You will therefore investigate the energetic and electronic characteristics of several possible surface orientations as a function of the polytype stacking sequence. Of particular interest is the position of the valence and conduction band with respect to the vacuum level, as this determines the redox activity of the surface. Anisotropic electron or hole mobilities would be valuable too, since this gives further information on the in- and out-of-plane conductivity of the new crystal structures. Finally, a good energetic description is possible by means of the axial next-nearest-neighbour Ising model (ANNNI), which attributes specific energy values to different close-stacked planes depending on the stacking with respect to the neighbouring layers.
In a second stage of your thesis, you will also look at adsorption at silver polytype surfaces. You will consider several small molecules and identify the most favourable adsorption site on the perfect and defected silver surfaces, as well as the corresponding adsorption strength. Such information is crucial to get an idea about the selectivity of different chemical reactions on silver.
For all of these simulations, you will use density-functional theory (DFT), which is a quantum physical method that allows describing the behaviour of nuclei and electrons at the atomic scale. You can use available software packages for which there is extensive expertise at the Center for Molecular Modeling.
You will be actively coached to acquaint yourself with the required simulation techniques early in the thesis year. In addition, this work is inspired by experimental collaborators at KULeuven. Your work will take place in close contact with these partners.
Context for Engineering Physics students
Physics: use of and insight in quantum mechanical models for materials modelling
Engineering: investigation of materials properties for industrially relevant applications

- Study programmeMaster of Science in Engineering Physics [EMPHYS], Master of Science in Sustainable Materials Engineering [EMMAEN], Master of Science in Physics and Astronomy [CMFYST]ClustersFor Engineering Physics students, this thesis is closely related to the cluster(s) MODELING, MATERIALS, NANOKeywordssilver, polytype, surface, Adsorption, density-functional theoryRecommended coursesSimulations and Modeling for the Nanoscale, Computational Materials PhysicsReferences
I. Chakraborty et al., ‘Novel hexagonal polytypes of silver: growth, characterization and first-principles calculations’, J. Phys.: Condens. Matter 23, 325401 (2011). https://dx.doi.org/10.1088/0953-8984/23/32/325401.
I. Chakraborty et al., ‘A stable, quasi-2D modification of silver: optical, electronic, vibrational and mechanical properties, and first principles calculations’, J. Phys.: Condens. Matter 26, 025402 (2014). https://dx.doi.org/10.1088/0953-8984/26/2/025402.
S. De Waele et al., ‘Error estimates for density-functional theory predictions of surface energy and work function’, Phys. Rev. B 94, 235418 (2016). http://dx.doi.org/10.1103/PhysRevB.94.235418.
Contact
Veronique Van Speybroeck